
un tratamiento médico?
Según la agencia francesa del medicamento ANSM, el desarrollo por un laboratorio de una molécula hasta la comercialización del medicamento necesita entre diez y quince años de investigación para explorar todos los aspectos
El desarrollo por un laboratorio de una molécula hasta la comercialización del medicamento necesita entre diez y quince años de investigación para explorar todos los aspectos.
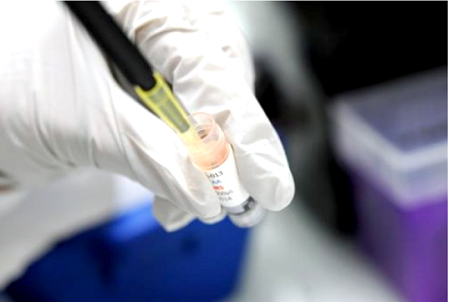
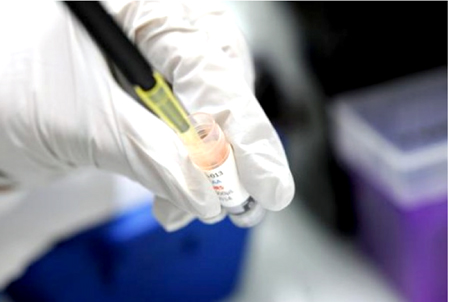
Al aprobar el empleo de tratamientos no homologados en la lucha contra la fiebre hemorrágica Ébola, la Organización mundial de la salud (OMS) decidió cortocircuitar un procedimiento de instalación en el mercado que puede demorar años.
Según la agencia francesa del medicamento ANSM, el desarrollo por un laboratorio de una molécula hasta la comercialización del medicamento necesita entre diez y quince años de investigación para explorar todos los aspectos.
Tras estudios experimentales en modelos animales o celulares, el laboratorio solicita la autorización de las autoridades sanitarias antes de lanzarse en un ensayo clínico en seres humanos (o estudio clínico) destinado a evaluar la eficacia y la tolerancia de un tratamiento.
El ensayo se desarrolla normalmente en tres etapas:
– la fase 1 permite evaluar la tolerancia y la ausencia de efectos indeseables en un pequeño grupo de voluntarios sanos (en general menos de 100).
– la fase 2 permite estimar la eficacia de la molécula y determinar su dosis óptima. Se realiza por lo general en unos pocos cientos de personas.
– la fase 3 compara el tratamiento a un placebo o a un tratamiento de referencia ante unas mil personas. El objetivo es mostrar la eficacia y evaluar la relación eficacia/tolerancia.
Tras los ensayos clínicos, que pueden durar entre cinco y seis semanas, el laboratorio dirige una solicitud de autorización de puesta en el mercado a la autoridad sanitaria competente. En Europa, se trata por lo general de la Agencia europea de medicamentos (EMA), mientras que en Estados Unidos, el laboratorio debe primero dirigirse a la Food and Drug Administration (FDA).
Para obtener esa autorización, el nuevo tratamiento debe en principio presentar una relación beneficio/riesgo al menos equivalente al de los productos ya comercializados.
Si ningún tratamiento está disponible, el procedimiento de autorización puede acelerarse «por razones compasivas»: por ejemplo en el caso de las «autorizaciones temporarias de utilización» otorgadas en Francia para permitir a ciertos enfermos en fase terminal utilizar medicamentos que no fueron aún puestos en el mercado.
Tras su comercialización, el medicamento permaneció bajo vigilancia, con una evaluación permanente de los efectos indeseables conocidos o nuevamente identificados. En caso de riesgo para la salud, puede ser retirado del mercado.
AFP









{kind=link}